By Xiaoming Xu, Ph.D., U.S. Food and Drug Administration
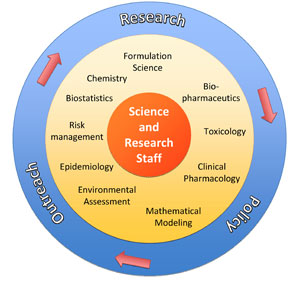
Science and Research Staff (SRS) is a science policy support group located within the immediate office of the Office of Pharmaceutical Sciences (OPS), in FDA’s Center for Drug Evaluation and Research (CDER). The group was established in 2004 and has since addressed a number of regulatory science questions and concerns by proactively conducting research and by contributing to the development of policies. Currently, SRS is engaged in activities including nanotechnology, bioequivalence of generic drugs, adverse drug report analysis, quantitative structure activity relationship (QSAR) modeling for the development of predictive models for QT prolongation, phospholipidosis, ecological toxicity and endocrine disruption, safety assessment of botanical drugs, dietary supplements, and compendial articles, environmental assessment of pharmaceutical drugs, and policy development and policy revisions regarding requirements for environmental assessment. For example, SRS was the first group within FDA to develop a manual of policies and procedures (MAPP) in the area of nanotechnology. The SRS mission is to provide both proactive and rapid response support to CDER in the development and implementation of regulatory science policies aimed at assessing and improving drug quality, safety, and efficacy, using a multidisciplinary “think tank” approach that utilizes internal resources, external collaborators, and contractors, to meet CDER priorities and goals.
Below is an interview with Dr. Nakissa Sadrieh, Associate Director for Research Policy and Implementation in OPS, CDER, FDA, and head of the Science and Research Staff.
Q: Can you give examples of types of activities conducted in the SRS?
A: The SRS conducts policy support activities through regulatory research. The group proactively identifies questions that pose tangible regulatory challenges, such as impeding regulatory decision making. The specific types of activities include 1) researching the CDER database of completed reviews to evaluate the data submitted by drug companies, 2) reviewing the adverse events databases and drug usage data to evaluate the impact and magnitude of the potential issue, 3) designing a research hypothesis that can be tested, 4) identifying collaborators and conducting the necessary research, and 5) publishing and disseminating the results of the work. Areas of interest currently include nanotechnology, safety and quality of generic drugs, drug quality research, use of mathematical modeling to develop predictive tools for preclinical safety assessment, and environmental assessment of pharmaceuticals.
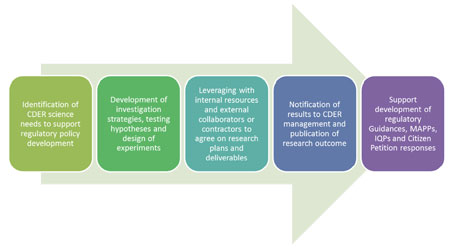 |
|
SRS model for supporting CDER regulatory science policy development (IQP stands for internal quality procedure) |
Q: What is the operating model at the SRS?
A: The SRS utilizes a unique model where the focus of the work is not only to conduct sound science but to ensure that the work has regulatory relevance and that it will impact science policy development. We call it the “three-pronged approach”: 1) research, 2) policy, and 3) outreach. During the research phase, the first step is to identify the scientific question that will address the regulatory policy gap, the next step is to develop a testable hypothesis for the research project, the third step is to identify collaborators, and the last step is to conduct the experimentation. During the policy phase, the first step is to frame the results of the research in such a way as to be able to address policy development or revision, and the second step is to help support and implement any policy-related activities. During the outreach phase, the goal is to publish and present the work both internally and externally, with the ultimate goal of impacting policy development.
|
|
|
The three-pronged approach operating model at SRS: research, policy, and outreach |
Q: How do you choose a project?
A: Projects are chosen because they will have the potential to impact policy development or revision of existing policies. Policy outcomes may result in the development or revision of guidance documents, development or revision of MAPP, and supporting agency responses to citizen petitions.
Q: How are research projects prioritized in your group?
A: Projects are prioritized based on their regulatory impact. For example, pharmaceutical drug product quality is an area of priority for CDER; therefore, one of our projects is aimed at minimizing the likelihood of glass delamination in injectable formulations. We are collaborating with experts in glass delamination, and the goal is to conduct research encompassing a risk management approach to support any necessary guidance development in order to decrease the formation of glass lamellae in drug formulations.
Q: Can you elaborate on your research project on generic antiepileptics?
A: We recently awarded a five-year contract with a ceiling of $5 million to conduct bioequivalence studies on generics that might have been associated with adverse events signals, such as toxicity or lack of efficacy. This is a continuation of our efforts to address concerns of neurologists and their patients regarding the bioequivalence of generic antiepileptic medications.
Q: Nanotechnology-based pharmaceutical drug products are emerging rapidly. What has the SRS done, or is currently doing, to understand how to better regulate such products?
|
|
|
Science and Research Staff group members. |

A: SRS has been working for years to identify issues that might present obstacles to the regulation of nanotechnology-containing drugs. Characterization of nanomaterials has been one of the biggest challenges in research as well as in drug development. Therefore, one of the main contributions of SRS has been to develop a MAPP to help identify those products that might contain nanoscale materials (CDER 2010). With this MAPP, SRS has been able to create an internal database of approved and under-review drugs that contain nanoscale materials. The results of the internal database analysis were presented at the August 2012 meeting of the Advisory Committee for Pharmaceutical Science (Sadrieh 2012). Additionally, preliminary analysis of the CDER nanotechnology database has formed the foundation for future CDER initiatives, such as the CDER risk assessment approach to the assessment of safety, efficacy, and quality of nanoscale materials in drug products, which was recently accepted for publication in the Nanotechnology in Drug Development theme issue of the AAPS Journal (Morefield et al. 2013).
Q: The SRS is also responsible for CDER’s environmental assessment (EA) review function. What projects other than review does the EA group engage in?
A: As part of the drug review process, pharmaceutical companies must conduct an EA of drugs, unless they qualify for a categorical exclusion. One of the projects within the SRS is to identify ways to improve the regulatory review of pharmaceuticals, as it relates to the EA process. As such, we are developing tools to help predict if certain drugs might have an ecotoxicologic effects, so that policies related to categorical exclusion might be improved. Another EA project revolves around better understanding characteristics of drugs known to be ecotoxic, especially when they have environmental effects at low concentrations. These responses at low doses are called nonmonotonic since they differ from the traditional models used in toxicology. One of the identified characteristics of nonmonotonic dose responses is associated with the ecotoxicity of hormonally active drugs, also called endocrine disruptors.
Q: What types of mechanisms does SRS use to accomplish its mission?
A: The SRS uses mechanisms such as contracts, research collaborative agreements (RCAs), memoranda of understanding (MOUs), and interagency agreements (IAGs). There are also laboratories that SRS collaborates with, both within CDER as well as in other FDA centers such as the National Center for Toxicology Research (NCTR). The use of IAGs and MOUs supports collaborations with other government agencies such as the National Institutes of Health (NIH) and National Institutes of Standards and Technologies (NIST). Finally, SRS uses contracts and RCAs with universities, software developers, consultants, and pharmaceutical companies.
References
CDER. 2010. Reporting format for nanotechnology-related information in CMC review, CDER MAPP 5015.9
Sadrieh, N. 2012. Overview of CDER experience with nanotechnology-related drugs
Morefield, E, Cruz, C, Tyner, K, Velazquez, L, Hyams, K, Jacobs, A, Shaw, A, Jiang, W, Lionberger, R, Hinderling, P, Kong, Y, Brown, P, Ghosh, T, Strasinger, C, Suarez, S, Henry, D, Vanuitert, M, and Sadrieh, N. CDER risk assessment exercise to evaluate potential risks from the use of nanomaterials in drug products, AAPS J. (in press, 2013).
More Related Links
FDA Guidance on Nanotechnology, www.fda.gov/ScienceResearch/SpecialTopics/Nanotechnology/default.htm
FDA CDER Science and Research Programs, www.fda.gov/Drugs/ScienceResearch/ucm319528.htm
This article is in the public domain and not copyrightable. It may be freely reprinted with customary crediting of the source. Controlled Release Society, 2013.

