This first wave of computational modeling in pharmaceutical science was the application of informatics-based methods to finding optimum drug molecule structures: drug design. We can now move beyond this, both from drug design to the design of drug delivery vehicles, nanomedicine, and from raw optimization to a tool capable of obtaining insight that will, in combination with complementary experimental methodologies, allow for such vehicles to be created through a rational design process akin to engineering machines. Our work on applying computational modeling to determine aspects of the structure and behavior in the bloodstream of liposome-based drug delivery systems (LDS) represents a case study in how this can be achieved.
Through nanomedicine, the development of nanoscale drug carriers known as nanoparticles (NPs), lies the promise of delivery of drugs with vastly decreased toxicity and increased efficacy.1 While there are several different forms of NPs, including polymeric micelles, solid NPs, dendrimers, and LDS, they all possess a common set of functional elements: 1) a core compartment where the drug to be delivered is stored, 2) a surrounding material that encapsulates the drug, 3) a protective sheath on the NP exterior that inhibits uptake by the mononuclear phagocyte system (MPS), and 4) possibly targeting moieties on the NP exterior to achieve active targeting. We are left with a wide array of metaphorical dials and switches to design NPs for optimized function. This includes the overall size of the NP and the formulation (molecules of which it is composed) of each functional element.
While the number of input parameters at our disposal in NP design is considerable, linking them to the structure and function of the NP is, however, not trivial; several factors beyond our control determine this, and our ability to elucidate the structure and function of NPs through experimental means alone thus remains limited: for the most part, NP design has been carried out through mostly trial and error based methodologies. We would argue that this is an important factor in why this field has, so far, been much more successful at generating publications than new drug therapies.2 Many experimental methodologies exist to study aspects of NP structure and function, each capable of monitoring a different separate aspect of the system; however, the collective insight is still incomplete. The situation is comparable to the parable of the blind men and the elephant: one grabs the trunk and thinks they touch a snake, another the tail and believes it to be a rope, another the leg and believes it a tree, and so on. Computational modeling using the multiscale modeling paradigm3 is just the tool capable of filling in the gaps and revealing the elephant.
When a NP is in the bloodstream its structure and behavior are manifested over a broad range of length and time scales. No single technique can hope to model all of this at once. Instead, several different models with different assumptions are used in conjunction, each one suited to a different range of length and time scales. At the bottom, covering the smallest scale, is molecular dynamics (MD) modeling with all-atom resolution. The real interactions between the atoms and molecules, on a fundamental level governed by quantum mechanics, can be approximated through a classical model: molecules as assemblies of metaphorical solid balls connected by springs and hinges that in turn collide with one another, through nonbonded interactions between all atoms. The result is what has been described by Klaus Schulten and coworkers as a “computational microscope,”4 essentially a holographic movie of the system with all-atom resolution and length and time scales of around 10–20 nm and around 0.5–1 μs, respectively. To reach larger length and time scales, this metaphorical microscope can be refocused through coarse graining: models with particles that are not single atoms but rather groups of atoms or even larger structures,3 for example, the MARTINI5 force field or the even more coarse-grained dissipative particle dynamics.3 Finally, at the top end are continuum models, with parameters determined by smaller-scale simulations, capable of reaching up to the macroscopic scale.
Our work, carried out over the past eight years, using molecular dynamics modeling with all-atom resolution to study LDS-based drug therapies, can be seen as a case study in how computational modeling can be used as a design tool in nanomedicine. In our work we have provided insight into four main areas: 1) the protective poly(ethylene glycol) (PEG) sheath (PEGylation), 2) targeting ligand design, 3) membrane formulation, and 4) interaction between the membrane and drugs carried by the LDS. Our work is described in detail in our recently published comprehensive review paper6 and two more recent publications.7,8 We will merely outline our work here and refer our readers to these three cited papers for a more complete understanding of what we have achieved.
The first issue we approached was the behavior of the PEG polymer coating of the LDS: PEGylation. The motivation for this research is that, although PEG is effective as

a protective polymer corona, there is significant room for improvement, and the search for alternatives is an active field of research. We started from the simplest models of a gel and liquid crystalline membrane with and without PEGylation and then added in levels of complexity one by one: varying PEG concentration and inserting cholesterol into the membrane to duplicate the formulation of Doxil® , 9 the first approved PEGylated liposome-based therapy (Figure 1). We looked at the behavior in plasma though inclusion of NaCl at a level of 125 mM and also investigated KCl and CaCl2 at the same ionic strength.
We found behavior that should have been expected, given what was previously known about PEG as a polymer on its own, but that in the context of use in LDS-based therapies was unanticipated. Because PEG is a polymer electrolyte it associates strongly with the Na+ ions. Also, PEG is known to be soluble in a wide range of both polar and nonpolar solvents and as a result entered into the membrane core of the more loosely structured liquid crystalline membrane but did not for the case of the gel membrane. When we simulated the Doxil® formulation9 with cholesterol in the membrane, the PEG entered into the membrane but in a specific fashion: winding along the β surface of the cholesterol molecules, thus disrupting the role they normally play in condensing the membrane. The density of the PEG layer was found to have an important effect: at 5% formulation density of PEGylated lipids the Cl– ions, with their tightly bound water shells, were found to sit in water pockets in the PEG layer. When the formulation density of PEGylated lipids was raised to 10% the Cl– ions and their tightly bound water shells were expelled from the PEG layer. Thus, as the formulation density of PEGylated lipids increases, the effective surface charge of the liposome, positive in the absence of a PEG corona due to Na+ ions bound to the lipid headgroups, initially decreases; however, when the formulation density of PEGylated lipids is increased beyond a certain density, the surface charge starts increasing again. Because surface charge plays

an important role in MPS uptake, this could explain the previously observed optimum PEGylated lipid density. It was also found that the divalent Ca2+ ions did not bind to PEG at all, preferring to bind to the lipid headgroups. It has previously been observed that PEGylation inhibits calcium-induced liposome fusion via headgroup crosslinking, and now this provides a mechanism: steric hindrance.
Active targeting design currently involves a significant degree of trial and error; ligands that yield positive results in isolated docking to their targets often fail in vivo when attached to the surface of the liposome. We have demonstrated that computational modeling has the capacity to yield significant insight here. In one example we studied a new targeting ligand, the activated endothelium targeting peptide (AETP) moiety, that was more hydrophobic than a similar ligand, the arginyl-glycyl-aspartic acid (RGD) peptide (Figure 2), which had previously been shown to work. Although the AETP moiety passed phage display screening it failed in vivo. Our computational modeling showed the cause of this failure: it was not, as previously suspected, that the ligand entered into the membrane core, but rather it was obscured by the PEG polymer itself. Thus, we could suggest that replacing the PEG polymer with a possibly more hydrophilic polymer could solve this problem. We also worked with an Indian research group developing LDS-based therapies to target liver cancer and showed with our simulations that exposure to the solvent was increased by polymerization of the ligand.7
In other work we investigated the structure of lipid membranes composed of synthetic lipids, for example, the membrane of a liposome proposed by the Szoka group with the phosphate and choline groups switched. Although this seems like a trivial change, we determined that the result significantly altered membrane properties: the membrane no longer binds Na+ ions, and the water ordering at the membrane surface is reversed, a feature that possibly aids drug delivery through the cell membrane. We have also studied the behavior of drugs being delivered by PEGylated
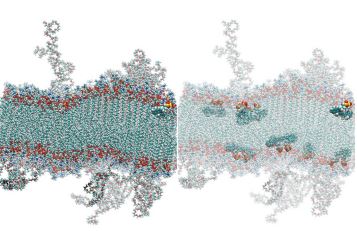
liposomes, for example, indocyanine green (Figure 3), and found that hydrophobic drugs can sit either in the membrane or the PEG layer; however, where they locate is determined by their starting point in the simulation.8 This indicates the possibility that the location of drugs in the liposome can be controlled through formulation.
Our work in this area continues; we are currently working on improving the existing coarse-grained force fields to allow us to investigate larger-scale properties of LDS-based delivery systems and ultimately use the insight from these simulations to construct continuum models capable of studying the entire LDS in the bloodstream. Our intention is ultimately to present our methodology as a novel framework in which computational modeling is applied in concert with complementary experiments to build the next generation of NPs using a rational design approach: as nanoscale engineered machines, or, to use the colloquial term, nanobots.
References
1. Duncan, R, Gaspar, R. Nanomedicine under the microscope. Mol. Pharm. 8(6):2101-2141 (2011).
2. Venditto, VJ, Szoka, FC. Cancer nanomedicines: So many papers and so few drugs! Adv. Drug. Deliv. Rev. 65(1):80-88 (2013).
3. Murtola, T, Bunker, A, Vattulainen, I, Deserno, M, Karttunen, M. Multiscale modeling of emergent materials: Biological and soft matter. Phys. Chem. Chem. Phys. 11:1869-1892 (2009).
4. Lee, EH, Hsin, J, Sotomayor, M, Comellas, G, Schulten, K. Discovery through the computational microscope. Structure 17:1295-1306 (2009).
5. Marrink, SJ, Risselada, HJ, Yefimov, S, Tieleman, DP, De Vries, AH. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111:7812-7824 (2007).
6. Bunker, A, Magarkar, A, Viitala, T. Rational design of liposomal drug delivery systems, a review: Combining insight from experimental and computational study of lipid membranes, liposomes and their PEGylation. Biochim. Biophys. Acta 1858(10):2334-2352 (2016).
7. Pathak, P, Dhawan, V, Magarkar, A, Danne, R, Govindarajan, S, Ghosh, S, Steiniger, F, Chaudhari, P, Gopal, V, Bunker, A, Róg, T, Fahr, A, Nagarsenker, M. Design of cholesterol arabinogalactan anchored liposomes for asialoglycoprotein receptor mediated targeting to hepatocellular carcinoma: In silico modelling, in vitro and in vivo evaluation. Int. J. Pharm. 509(1-2):149-158 (2016).
8. Lajunen, T, Kontturi, LS, Viitala, L, Manna, M, Cramariuc, O, Róg, T, Bunker, A, Laaksonen, T, Viitala, T, Murtomäki, L, Urtti, A. Indocyanine green loaded liposomes for light triggered drug release. Mol. Pharm 13(6):2095-2107 (2016).
9. Gabizon, A, Catane, R, Uziley, B, Kaufman, B, Safra, T, Cohen, R, Martin, F, Huang, A, Barenholz, T. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 54(4):987-992 (1994).

